

Diasorin (FTSE MIB: DIA) announces that it has received 510(k) clearance from the U.S. Food and Drug Administration (FDA) for the company’s NxTAG® Respiratory Pathogen Panel (RPP) v2. This updated panel, an addition to Diasorin’s expanding molecular multiplexing portfolio, responds to customer needs by enhancing test usability.
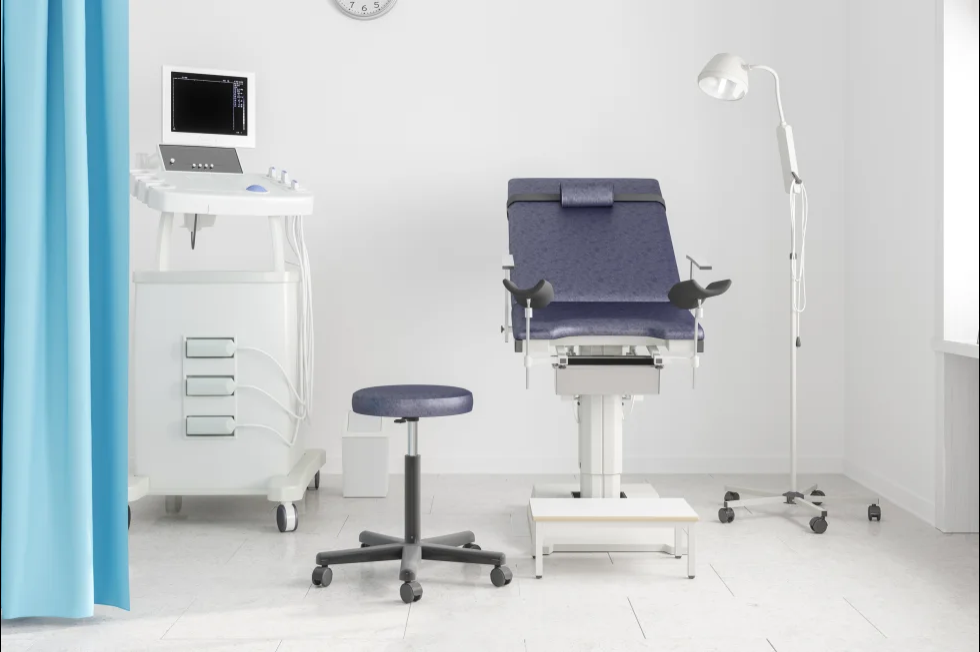
Instead of a traditional speculum-involved pelvic exam to screen for cervical cancer, the US Food and Drug Administration has given the go-ahead for patients to have the option to collect their own vaginal samples for screening in a health care setting, such as at their doctor’s office, an urgent care or even a mobile clinic.

Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today the FDA approval of its human papillomavirus (HPV) self-collection solution, one of the first available in the United States. Screening for HPV can help identify women who are at risk of developing cervical cancer so that the disease can be found and treated early before cervical cancer has a chance to develop.

The US Food and Drug Administration (FDA) has warned users not to use Cue Health’s at-home Covid-19 tests, just one week after the agency issued a warning letter saying that Cue was not following the conditions specified in the emergency use authorisations for its tests.

QIAGEN (NYSE: QGEN; Frankfurt Prime Standard: QIA) today announced that the U.S. Food and Drug Administration (FDA) has cleared the QIAstat-Dx Respiratory Panel Plus syndromic test for clinical use.

Stay up-to-date with the latest happenings in the rapidly evolving field of In Vitro Diagnostics (IVD) in China.

The US Food and Drug Administration released on Monday its final rule to regulate laboratory-developed tests (LDTs).

Recently, Wondfo Biotech's wholly-owned subsidiary in the United States, Wondfo USA Co., Ltd. (hereinafter referred to as the " Wondfo USA"), received notification from the U.S. Food and Drug Administration (FDA) that the WELLlife™ COVID-19/Influenza A&B Test has been granted Emergency Use Authorization (EUA240004). The WELLlife™ COVID-19/Influenza A&B Test is designed for professional use at the point-of-care (POC) settings.

Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that its Elecsys pTau217 assay received Breakthrough Device Designation from the U.S. Food and Drug Administration (FDA). This blood test, which is being developed in collaboration with Eli Lilly and Company, will be used to help identify the presence or absence of amyloid pathology in individuals, which can help ensure they are able to receive appropriate care. This may include participation in clinical trials or access to approved disease-modifying therapies. If approved, the test could help rapidly broaden access to a more timely and accurate diagnosis and potentially mitigate the impact of Alzheimer’s disease on people and society.

Labcorp (NYSE: LH), a global leader of innovative and comprehensive laboratory services, announced today the U.S. Food and Drug Administration (FDA) has granted Emergency Use Authorization (EUA) for its Mpox PCR Test Home Collection Kit to aid in the diagnosis of infection with non-variola Orthopoxvirus, including the monkeypox virus that causes monkeypox, also known as mpox. The test is the first mpox at-home collection kit authorized by FDA and is available to physicians to order for patients 18 years of age or older who are suspected of mpox infection.
✔ All (388)
✔ Press release (3)
✔ Industry news (385)
Don’t miss important updates about the show and the in vitro diagnostic industry.
Sign-up for our newsletter today.





Copyright © 2024 GL events Ruihe (Shanghai) Exhibition Co., Ltd. All Rights Reserved. ( 沪ICP备12004745号-1 )

We deliver the latest IVD news straight to your inbox. Stay in touch with CACLP News.
sign-up for our newsletter today.



To ensure our newsletter hit your inbox, make sure to add @caclp.com to your safe senders list. And, as always, feel free to contact
us with any questions and thanks again for subscribing.
Go back







